Skinny Label Infringement: When Carve-Outs Fail and Royalty Streams Bleed

The "skinny label" is a regulatory permission slip to seek approval for fewer than all conditions of use of a reference product. In theory, it enables clean market segmentation: a generic enters for off-patent indications while the innovator's second medical use patent remains commercially intact. In practice, the construct sits atop prescribing systems that cannot enforce indication-level boundaries at the dispensing edge—and the resulting leakage propagates directly into royalty economics, damages models, and the underwriting assumptions of a $29.4 billion royalty financing market.
This analysis maps the failure mechanism from regulatory architecture through litigation doctrine to royalty finance—and situates it against the Supreme Court's upcoming oral arguments in Hikma v. Amarin (April 29, 2026), which could redefine when skinny labels trigger induced infringement liability.
The Regulatory Architecture: Formally Clean, Operationally Porous
In the United States, the mechanism sits inside the Hatch–Waxman patent certification architecture. An ANDA applicant may submit a section viii statement certifying that an Orange Book method-of-use patent "does not claim" the use for which approval is sought, rather than certifying against it. FDA's long-standing position is that a generic's label must generally match the reference listed drug, except for "permissible" differences—with carve-outs for patent- or exclusivity-protected conditions of use being the canonical example.
In the European Union, the legal hinge is explicit in the consolidated text of Directive 2001/83/EC: for generic authorisations under Article 10, the generic's SmPC need not include those parts of the reference SmPC that refer to indications or dosage forms still covered by patent law at the time the generic is marketed. The European construct is not merely "tolerant" of carve-outs; it contemplates them.
What matters for infringement risk is not the elegance of this segmentation on paper. The patent question is whether, downstream, the generic's conduct is best characterised as lawful sale into non-protected uses—with inevitable leakage treated as system noise—or a legally cognisable form of encouragement, targeting, or facilitation that triggers liability. The gulf between those narratives is where skinny labels fail.
Two administrative artefacts amplify the fragility. First, the U.S. Orange Book's use codes are not abstract academic metadata: they are the mechanism that connects a method-of-use patent to particular approved uses and are embedded in the data FDA publishes for Orange Book listings. Second, the EU's carve-out permission is coupled to national substitution and prescribing regimes, which are not harmonised as a single operational layer. Even within the EU, substitution and prescribing rules vary materially by Member State—mandatory INN prescribing in some countries (Greece, Portugal, Estonia), mandatory substitution in others (Finland, Sweden), and differing scope and implementation everywhere else.
Skinny labelling, in short, is a regulatory attempt to slice a single molecule's commercial life into patent-shaped tranches. In most healthcare systems, the molecule is the unit of prescribing and dispensing—not the tranche.
Cross-Use Economics: INN Prescribing and the Leakage Parameter
The practical failure mode is predictable. A molecule has at least two meaningful demand pools—call them A (off-patent) and B (patented second medical use). The generic launches with approval for A, but because clinical practice is molecule-centred, it gets dispensed for B as well.
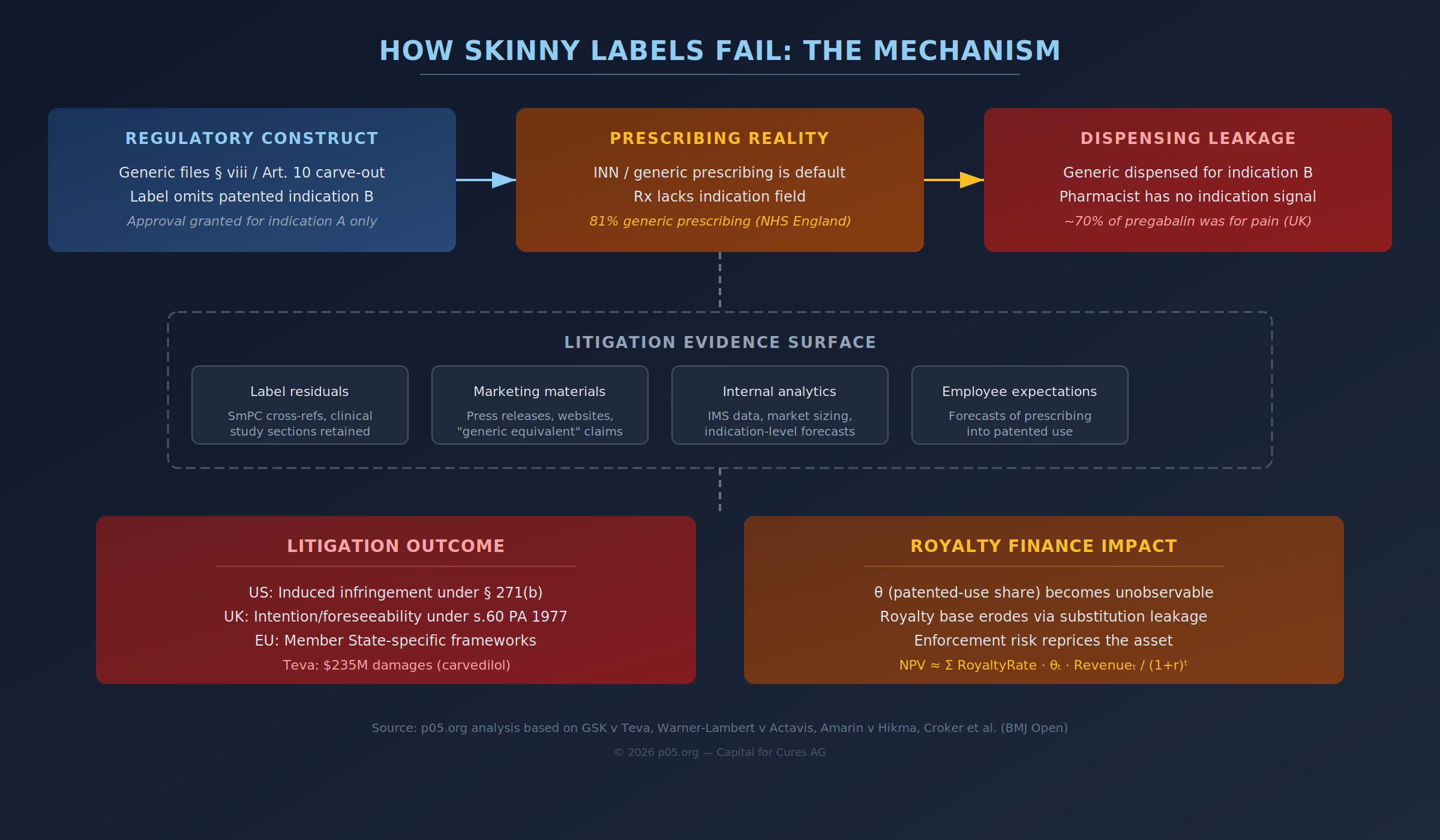
This is not an exotic edge case in jurisdictions with entrenched generic prescribing. In England, the NHS Business Services Authority reports that 81% of all drugs in primary care are prescribed generically, reflecting a deep systemic preference for INN prescribing. Across the EU, a 2025 survey-based analysis in the European Journal of Public Health describes INN prescribing and substitution rules as present in all Member States surveyed, with substantial variation in scope and implementation.
The technical point is not merely that generic prescribing exists. It is that the dispensing event often lacks an indication field robust enough to enforce indication-level patent segmentation at scale. In the UK pregabalin litigation record, Arnold J treated as important the operational reality that pharmacists are frequently presented with generic prescriptions that do not state the indication—which, unless additional steps are taken, makes it likely some generic product will be dispensed into the patented pain indication. Croker et al. (BMJ Open) explicitly note that the prescribing dataset they used does not include indication, constraining their ability to identify whether an individual generic pregabalin prescription related to neuropathic pain.
From an economic lens, this creates a leakage parameter that is both central and hard to observe. If total generic unit sales over a damages window are S_Total and the proportion economically attributable to the patented use is θ, then a stylised exposure model is:
Damages ≈ θ · S_Total · RoyaltyRate
The structure is clean; the estimator for θ is not. Even in a system with high-quality utilisation data, θ is typically inferred, not observed, and inference quality depends on whether claims or dispensing events cleanly map to diagnosis or indication. Croker et al. had to work around precisely this problem in the NHS setting, despite unusually rich national-level prescribing data.
This is where skinny labels become less a question of doctrinal purity and more a question of health-system information architecture. If your infrastructure cannot reliably express "this prescription is for B", then your patent enforcement strategy is forced into second-best proxies: guidance letters, software prompts, pharmacy notifications, contracting restrictions, and—ultimately—litigation narratives about intent.
The Infringement Theory: Inducement in the US and Intention Frameworks in Europe
In the United States, skinny-label disputes typically land in induced infringement under 35 U.S.C. § 271(b): "Whoever actively induces infringement of a patent shall be liable as an infringer." Standing alone, that sentence hides the real fight. Inducement requires a mental element. The Supreme Court in Global-Tech v SEB endorsed a knowledge-based standard and held that willful blindness can satisfy the knowledge requirement.
Operationally, this turns skinny-label litigation into an evidentiary contest about whether the generic merely sold a product that could be used in an infringing way, or whether it intentionally encouraged the infringing use and caused it. The Federal Circuit's GSK v Teva opinion makes causation explicit: to establish inducement, the patent owner must show that the accused inducer's actions actually induced the infringing acts of another and that the inducer knew or should have known the actions would induce infringement.
In the UK and parts of Europe, the doctrinal scaffolding looks different, but the conceptual tension is identical: second medical use protection collides with molecule-centric prescribing. UK second medical use claims (in the Swiss-form tradition) have been litigated through infringement provisions such as section 60 of the Patents Act 1977. The pregabalin Supreme Court judgment records disagreement within the court about whether subjective intent is relevant versus an objective "presentation" test—how the product is prepared, labelled, and put on the market.
For an expert audience, the commonality across jurisdictions is more important than the labels on the doctrines. Across both systems, the decisive facts often live outside the carved-out indication text: what the label still implies even after deletion; how the product is presented (SmPC/PIL, "blue box" wording, cross-references); how the company communicates with prescribers, procurement bodies, or pharmacies; and what internal documents show about expected utilisation by indication.
That last category is where modern analytics makes the law sharper. The more precisely a company forecasts leakage into the patented segment, the easier it becomes for an opposing party to argue "knowledge of infringement" or even "intent to capture" the protected market—in the U.S. sense of inducement, or in European debates about foreseeability and targeting.
Pregabalin and Lyrica: A Second Medical Use Patent Meets an INN-Based NHS
The pregabalin (Lyrica) litigation is cited so often because it forces the legal abstraction to confront the operational substrate. The UK Supreme Court records the core configuration: Actavis launched "Lecaent" pregabalin under a skinny label (epilepsy and generalised anxiety disorder), while neuropathic pain remained the economically critical, disputed indication.
The system response was not a neat compliance check against the SmPC. It became a national workflow intervention. NHS England issued formal guidance—explicitly stating it was required by the court—directing that when pregabalin is prescribed for neuropathic pain, it should, so far as reasonably possible, be prescribed and written by the brand name Lyrica rather than the generic name. This is an extraordinary attempt to retrofit indication-level patent boundaries onto a generic-prescribing system.
Croker et al. provide unusually quantitative evidence of what happened next. After the NHS England instruction:
| Metric | Before Directive | After Directive |
|---|---|---|
| Pregabalin Rx'd as Lyrica | 0.3% | 25.7% |
| Estimated share of pregabalin use for pain | ~70% | ~70% |
| Practices prescribing Lyrica at pain-consistent levels | — | 11.6% |
| Estimated NHS excess cost (to July 2017) | — | ~£502 million |
Source: Croker et al., BMJ Open
Two details from the High Court record illuminate why the problem is structural rather than moral. First, the generic's own internal segmentation by indication became part of the factual landscape. Arnold J records that Actavis analysed IMS data and concluded roughly 71% of the pregabalin market related to pain, with the remainder outside the patented use, and that Actavis prepared both skinny-label and full-label materials to enable rapid launch decisions. This is precisely the kind of "data exhaust" that modern commercial teams generate routinely—and the kind that, in litigation, can be reinterpreted as knowledge of the protected segment's size and the inevitability of leakage.
Second, the court treated as foreseeable that a skinny label alone would not prevent dispensing for pain. The judgment expressly notes Actavis' knowledge that, unless further steps were taken, it was likely some product would be dispensed for pain because pharmacists were presented with generic prescriptions that did not state the indication. That sentence is the entire skinny-label dilemma in one place: the label is carved out, but the system cannot consistently act on the carve-out at the dispensing edge.
In the Supreme Court's final resolution, validity drove the outcome—key claims failed for insufficiency, so infringement did not ultimately decide market access. But the doctrinal wrangling about infringement tests, and the operational reality revealed by the litigation, are the enduring lessons.
Carvedilol and Coreg: The $235 Million Proof That § viii Is Not a Safe Harbour
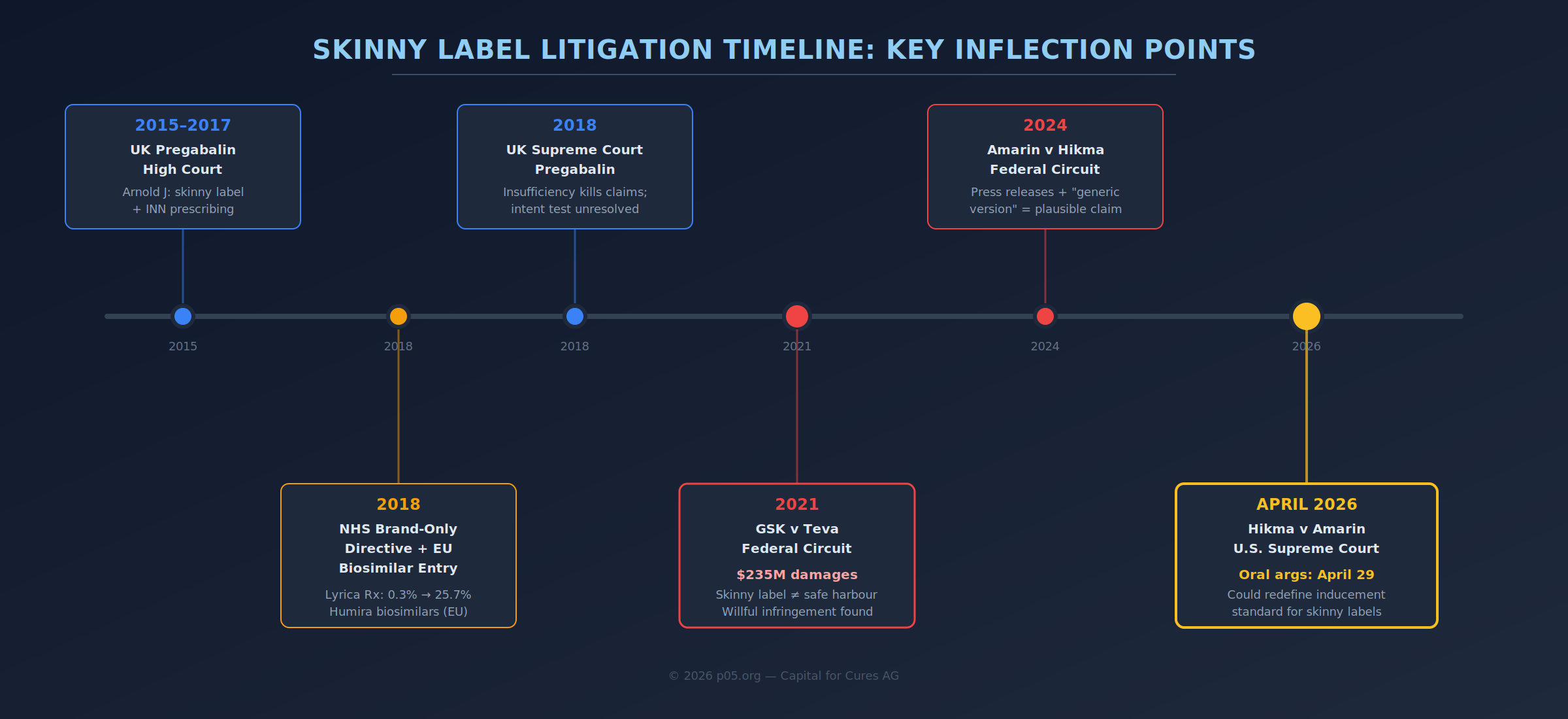
If pregabalin is the canonical NHS collision, GlaxoSmithKline v Teva is the canonical U.S. post-launch inducement collision. The Federal Circuit's 2021 per curiam opinion is explicit about the key timeline: Teva had a "partial label" period in which its label omitted the chronic heart failure indication, and a later "full label" period in which all indications appeared. The jury found induced infringement during both periods and found Teva's infringement willful, awarding $235 million in damages on a combination of lost profits and a reasonable royalty.
The critical point for skinny-label doctrine is what the Federal Circuit treated as "substantial evidence" supporting inducement despite the carve-out. The court summarised GSK's evidentiary theory in terms that should make any generic regulatory team uncomfortable: evidence that Teva's partial label did not successfully carve out the patented use, plus evidence that doctors read labels, that marketing materials guided doctors to the label and to Teva's website, that press releases encouraged prescribing for the patented use, and that Teva's own employees expected such prescribing.
Two doctrinal elements deserve emphasis because they structure the risk in a way financial models often underweight.
First, causation is not treated as an afterthought. The Federal Circuit framed inducement as requiring proof that the inducer's conduct actually caused physicians to infringe, and that circumstantial evidence could satisfy that requirement under the jury instructions. "Inevitable substitution" is not automatically "caused by the generic"—the case turns on whether the generic's communications and label content are found to have pushed the infringing use.
Second, the "label" in skinny-label litigation is rarely limited to the black-letter indication section. In GSK v Teva, the court's discussion does not read like a narrow parsing of excised text; it reads like a holistic assessment of product information flows—label content, catalogues, press releases, websites, and internal expectations. For firms that still treat section viii carving as primarily a redlining exercise managed by regulatory affairs, the case is an empirical warning: post-launch communications are part of the infringement surface.
The U.S. Supreme Court declined to review GSK v Teva in 2023, leaving the Federal Circuit's approach as governing precedent. For the market, the signal was straightforward: skinny labels are not a judicially guaranteed safe harbour.
Amarin v Hikma and the Vascepa Question: The Supreme Court Finally Weighs In
The Amarin v Hikma case has now escalated to the highest possible resolution point. On January 16, 2026, the Supreme Court granted certiorari, with oral arguments scheduled for April 29, 2026 and a decision expected before the Court's summer recess in early July.
The factual configuration is structurally illuminating. Amarin's Vascepa (icosapent ethyl) carries two indications: severe hypertriglyceridemia (SH), for which patents have expired, and cardiovascular risk reduction (CV), protected by method-of-use patents. The CV indication—approved in 2019—is where the vast majority of Vascepa's commercial value resides. Hikma filed a section viii carve-out, obtained approval solely for the SH indication, and launched with a skinny label in November 2020.
What makes this case different from GSK v Teva is the nature of the allegedly inducing conduct. Hikma's label itself contained no instruction to prescribe for the cardiovascular indication. Instead, Amarin's inducement theory rests on extra-label statements: press releases describing Hikma's product as the "generic version" of Vascepa, citations of Vascepa's total sales figures without distinguishing between indications, and website listings indicating therapeutic equivalence in a category broader than Hikma's approved indication.
The Federal Circuit reversed a district court dismissal in June 2024, holding that while the skinny label alone may not have encouraged infringing use, the totality of Hikma's statements plausibly stated a claim for induced infringement. The Trump administration's Solicitor General argued that the Federal Circuit ruling "subverts Congress's balance between competing interests" and supported Hikma's appeal.
The two questions presented to the Supreme Court are:
- Whether, when a generic drug label fully carves out a patented use, allegations that the generic drugmaker calls its product a "generic version" and cites public information about the branded drug are enough to plead induced infringement of the patented use.
- Whether a complaint states a claim for induced infringement of a patented method if it does not allege any instruction or other statement by the defendant that encourages, or even mentions, the patented use.
A ruling favouring Amarin could cement broad inducement liability for any marketing statements by generics that don't explicitly limit themselves to carved-out indications. A ruling favouring Hikma could reinforce the statutory carve-out pathway and reduce launch risk for generics and biosimilars. The legislative backdrop intensifies the stakes: the bipartisan Skinny Labels, Big Savings Act (S 5573), introduced in December 2024, proposes statutory safe harbour for skinny-label generics, but congressional action has not overtaken the Court's timeline.
For royalty investors, the pending decision is a binary event risk on any asset whose value story depends on second medical use patent enforcement.
Damages Attribution, θ, and the Hidden State Variable in Royalty Finance
For damages, the hard part is not writing the algebra; it is defending the attribution. Even in a single-payer environment with unusually transparent prescribing data, Croker et al. had to acknowledge that their dataset lacked indication. In the High Court pregabalin record, the absence of indication on prescriptions is treated as a direct driver of foreseeable dispensing into the patented use. The variable that damages models need most—θ, the patented-use share of sales—is often precisely the variable the dispensing infrastructure fails to express with certainty.
That uncertainty does not stay confined to courtrooms. It feeds directly into royalty economics and royalty-backed financing, where a second medical use patent frequently functions as a meaningful part of the asset's value story.
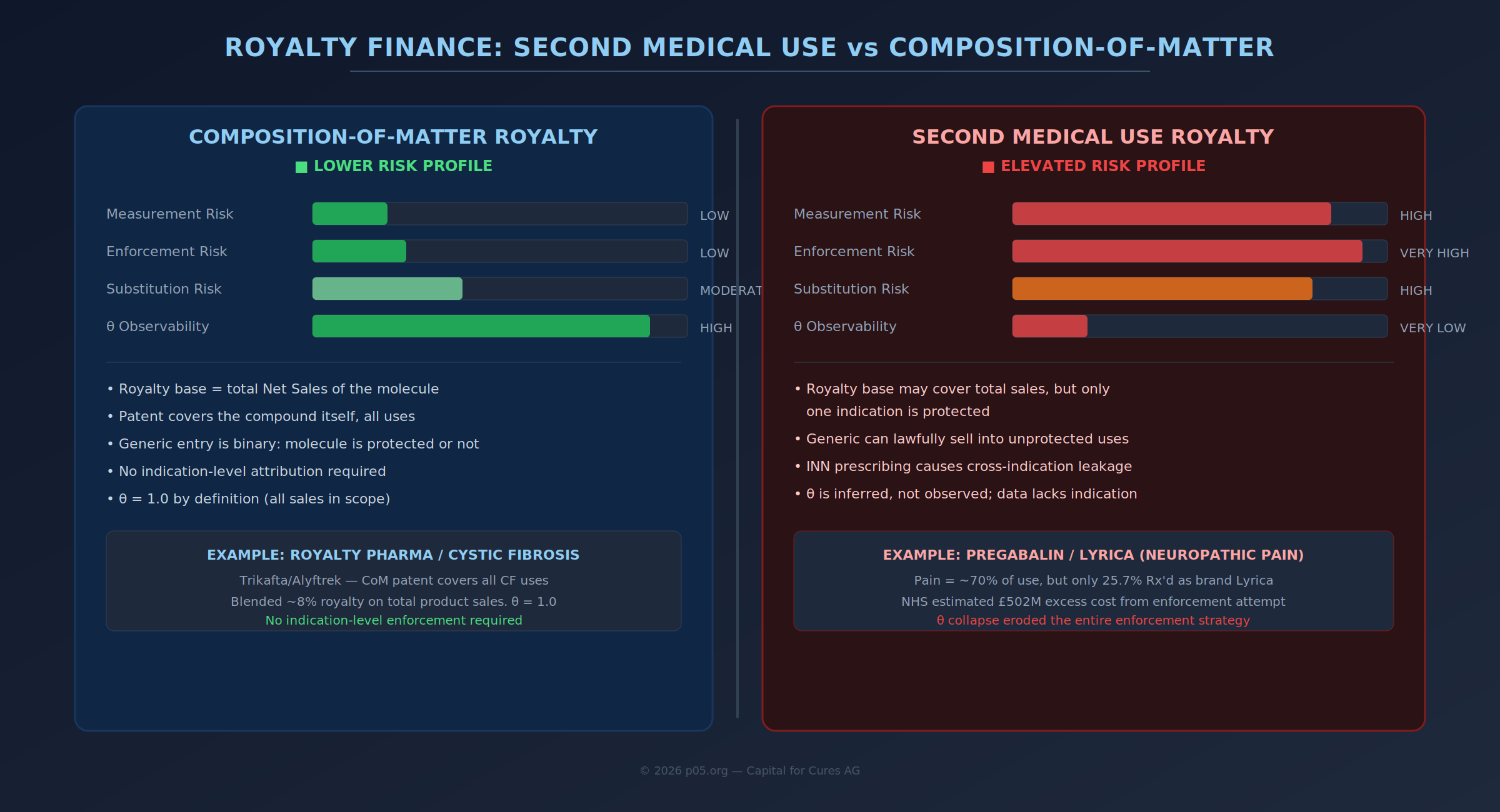
Royalty monetisation and royalty financing are now mainstream non-dilutive tools in life sciences capital formation. From 2020 through 2024, biopharma royalty financings totalled approximately $29.4 billion—more than double the amount raised between 2015 and 2019—with synthetic royalty structures growing at a 33% compound annual rate. The momentum continued into 2025, with Royalty Pharma alone executing transactions including a $2 billion arrangement with Revolution Medicines, an $885 million deal for Amgen's Imdelltra, $500 million for Teva's anti-IL-15 antibody, and multiple deals in the $250–350 million range. European activity is also accelerating—Goodwin recently advised on deals with GENFIT (France), Heidelberg Pharma (Germany), and MorphoSys (Germany).
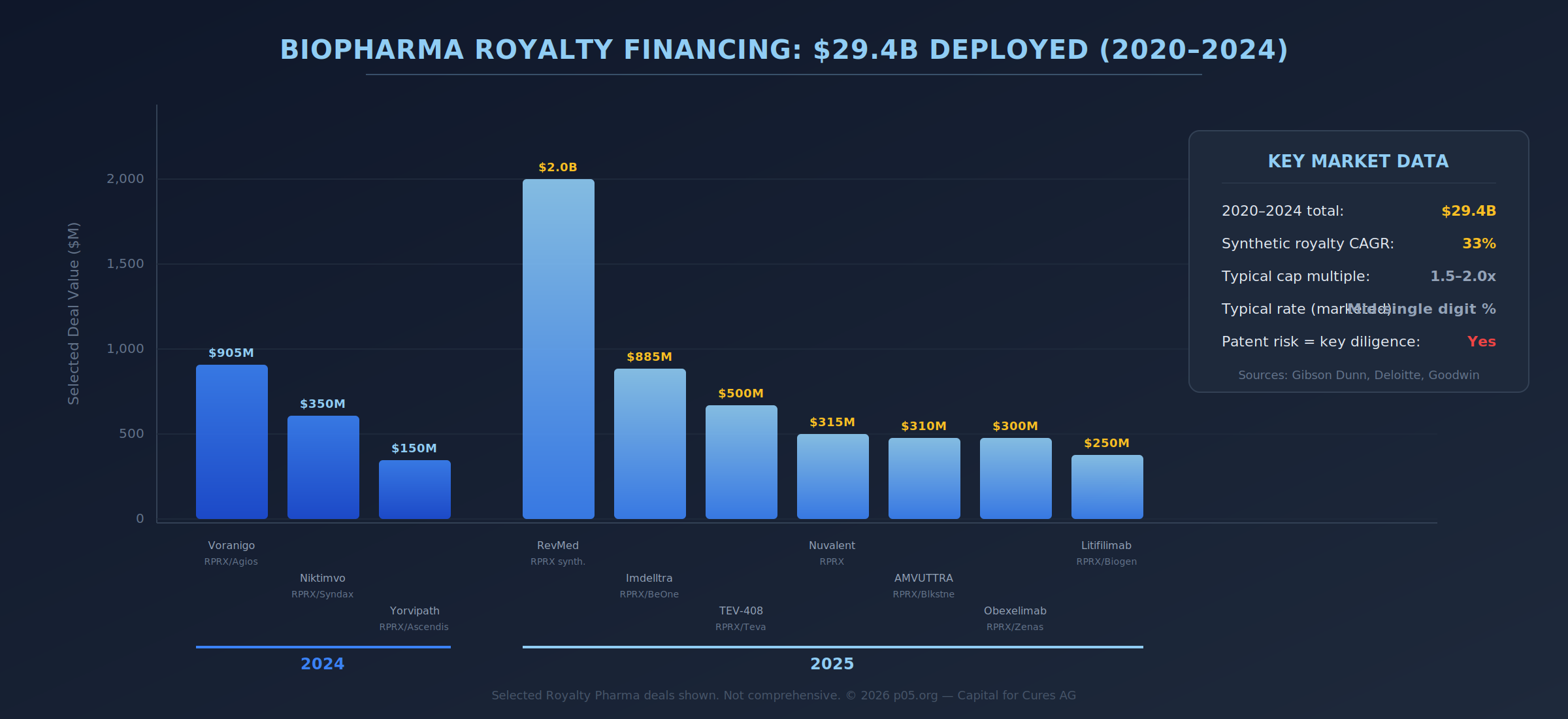
Structurally, a royalty monetisation investor purchases the right to receive a future stream of payments—a receivable-like asset—in exchange for upfront capital and an expected return profile. The typical synthetic royalty rate sits in the mid-single digits, with caps commonly in the range of 1.5–2.0x invested capital over a five- to eight-year horizon. Patent durability is the central underwriting variable: purchasers focus intense diligence on the ability of subject patents to survive challenge, both in federal court and before the USPTO.
Now place a skinny-label failure mode inside this royalty model.
If the protected indication represents a revenue fraction, and the royalty stream's present value is:
NPV ≈ Σ RoyaltyRate · θₜ · Revenueₜ / (1+r)ᵗ
then the entire edifice depends on what happens to θ over the royalty period. In many licensing and royalty agreements, the legal royalty base may be defined as total product "Net Sales" in a territory, even if the economic justification for the royalty is a second-use patent. That contractual mismatch becomes acute when θ collapses because enforcement fails, substitution regimes overwhelm the attempt at segmentation, or courts narrow inducement and intent theories.
This is why second medical use royalties are disproportionately exposed to measurement risk and enforcement risk compared with composition-of-matter royalties. Practitioners explicitly warn that royalty provisions become complex when the patents being licensed contain second medical use claims, because the contract must reflect the parties' intentions as to what sales are in-scope, and how disputes over scope will be handled. Patent licensing practice recognises the need for territory- and expiry-linked adjustments—step-downs when patent coverage lapses, for instance—to avoid misalignment between legal coverage and commercial payments.
Adalimumab and the Biosimilar Extension: Patent Thickets, Royalty Settlements, and Indication Carve-Outs at Scale
The Humira (adalimumab) ecosystem illustrates how indication-level patent strategies interact with royalty economics at unprecedented scale. AbbVie built a patent thicket of over 240 patents around Humira, and when eight biosimilar companies challenged them, the resolution took the form of global settlement agreements with a consistent architecture: biosimilars could enter the EU market from October 2018 but were delayed from the U.S. market until 2023, with each biosimilar manufacturer paying undisclosed royalties to AbbVie for licensing its Humira patents.
The biosimilar entrants that launched in Europe before 2023 often did so with skinny labels—carving out indications still protected by method-of-use or orphan drug exclusivity. A 2023 analysis in JAMA Internal Medicine estimated that Medicare savings from skinny-label biosimilars between 2015 and 2020 amounted to approximately $1.5 billion, with future savings anticipated from adalimumab biosimilars in particular.
But the demand-shaping mechanism for biologics is not retail pharmacy substitution; it is institutional switching and procurement. NHS England's commissioning framework for "best value biological medicines" is premised on early adoption and widespread use of biosimilars to free resources and support access. At the procurement layer, tenders for biologic and biosimilar medicines are routinely issued as large, centralised supply contracts with award criteria heavily weighted to price—an illustration of molecule- and contract-centred purchasing rather than indication-specific segmentation.
Interchangeability adds another jurisdictional split. In the EU, EMA and Heads of Medicines Agencies issued a joint statement supporting biosimilar interchangeability while noting that each Member State decides how switching and substitution are applied in its territory. In the U.S., FDA operates a distinct "interchangeability" designation pathway. For royalty underwriting, this means substitution velocity is not a single scalar—it is an interaction between regulatory status, payer and procurement incentives, and local switching rules.
The rate of skinny-label usage itself appears to be declining under litigation pressure. Between 2015 and 2019, roughly half of new generic prescriptions for brand-name drugs with use patents entered the market with skinny labels. Recent data shows annual declines, with a notable dip in 2023 following the GSK v Teva decision—a potential signal that fewer generic and biosimilar manufacturers are willing to use the pathway while legal uncertainty persists.
The AI and Real-World Data Angle: Discovery Risk Gets Sharper
The "AI and real-world data" angle is not speculative; it is already visible in the litigation record. The High Court pregabalin judgment records indication-level market sizing based on IMS data and internal planning around what share of the market is pain versus non-pain. In GSK v Teva, the Federal Circuit treated internal expectations—employees expecting prescribing for patented uses—as part of the inducement evidence mosaic.
As internal analytics become more sophisticated—segmenting by patient cohort, clinical pathway, prescriber type, tender outcome, and switching probability—the probability increases that discovery will surface documents that look, to a claimant, like contemporaneous acknowledgement of leakage into the protected indication. Companies that use machine learning to predict prescribing patterns, monitor biosimilar uptake, or model indication-level market share are generating precisely the kind of evidence that, in a future litigation context, could be recharacterised as "knowledge of infringement" or "intent to capture" the protected market.
For generic and biosimilar manufacturers, this creates a paradox: the more analytically sophisticated your commercial operations, the larger the documentation footprint that could be used against you in an inducement action. For royalty investors performing diligence on second medical use assets, the question is not whether the licensee has good data—it is whether the licensee's data discipline extends to understanding how that data might appear in an adversarial discovery process.
Where the Doctrine and the Plumbing May Move
The most realistic future stabilisers are not purely doctrinal; they are infrastructural.
One route is better data representation—cross-border and national initiatives that standardise medicinal product identification and eDispensation records. The 2025 EU study on INN prescribing and substitution situates these rules inside the broader push toward IDMP-based ePrescribing, and notes that European Health Data Space-related requirements for ePrescription and eDispensation reporting become legal requirements in 2026. If indication fields become more consistently captured and interoperable (a non-trivial governance problem), θ becomes less inferential and enforcement becomes less reliant on blunt instruments like brand-only prescribing directives.
The other route is judicial pressure for "affirmative steps", implicitly forcing generics and biosimilars to operationalise carve-outs through communications and supply-chain controls. The UK pregabalin record shows the contours of that debate: courts examined what steps were foreseeable, what notifications could be sent, and how prescriptions without indication undermine carve-outs. The U.S. trajectory, as GSK v Teva and now Hikma v Amarin illustrate, is that "label carve-out" alone may not be dispositive where surrounding conduct is treated as encouragement and causation.
A third route is legislative. The Skinny Labels, Big Savings Act would create an explicit statutory safe harbour for generics that comply with FDA carve-out requirements and do not reference the patented indication in labeling, promotion, or marketing. The bill applies to both generic and biosimilar products and would extend protections to conduct occurring before, on, or after enactment. FDA itself has expressed sufficient concern about the chilling effects of the GSK v Teva decision to request congressional action clarifying the exception from patent infringement for generic manufacturers with a skinny label.
For innovators, generics, and royalty investors, the upshot is the same: skinny labels are not merely regulatory drafting exercises. They are fragile equilibrium devices sitting on top of prescribing systems that were not designed to respect indication-level patent segmentation. When the equilibrium breaks, the economic damage is mediated by an unobservable parameter—θ—and the legal damage is mediated by whatever the factual record can be made to imply about knowledge, intent, and causation.
The Supreme Court's decision in Hikma v Amarin, expected by July 2026, will determine whether the current equilibrium gets a stabilising floor—or whether the fragility gets codified as the law of the land.
This article is published for informational purposes only. The author is not a lawyer or financial adviser. Nothing in this article constitutes investment, legal, or regulatory advice. Readers should consult qualified professionals before making decisions based on information presented here.
Member discussion