The Surrogate Marker Paradox in Chronic Disease

Why Many Interventions Improve a Biomarker Without Altering the Underlying Condition
Surrogate Markers vs True Outcomes
Clinical research often relies on surrogate markers – laboratory values or imaging findings that stand in for actual clinical outcomes. Examples include tumor shrinkage or progression-free survival in cancer, blood glucose (HbA1c) levels in diabetes, and amyloid plaque load in Alzheimer’s disease. These markers are convenient and respond quicker to interventions than the hard endpoints like survival or functional status. However, a growing body of evidence shows that improving a surrogate does not always translate into meaningful patient benefit (pharmacytimes.com).
A comprehensive 2024 meta-analysis in JAMA found that most surrogate endpoints used for FDA approvals in non-oncologic chronic diseases “lacked high-strength evidence of associations with clinical outcomes.” In other words, many drugs won approval by changing a marker, yet subsequent studies failed to confirm a significant impact on how patients actually feel, function, or survive (eprints.gla.ac.uk). Similarly, a review of oncology trials found that surrogate endpoints like tumor response or progression-free survival had only weak correlations with overall survival in many cases.
Why does this happen? One reason is that disease processes are complex – focusing on a single biomarker may neglect other pathological pathways. Some surrogate markers are not causal drivers of disease but rather bystanders. Lowering or raising such a marker might not alter the true disease trajectory. In some cases, aggressively pushing a marker in one direction can even cause off-target harm, worsening the overall outcome.
Regulatory history provides stark examples: the U.S. FDA’s accelerated approval program allows drugs for serious diseases to be approved based on surrogate endpoints (e.g. tumor shrinkage), but it requires confirmatory trials later (pmc.ncbi.nlm.nih.gov). When those trials fail to show real clinical benefit, approvals can be withdrawn. This occurred with bevacizumab (Avastin) in metastatic breast cancer – it initially gained approval because it improved progression-free survival, but was later withdrawn when no overall survival benefit (and substantial toxicity) was demonstrated(pmc.ncbi.nlm.nih.gov). In general, treating a number on a lab test or scan is not enough; we must address the underlying biology of the disease. Recent research has zeroed in on one common biological thread across many chronic conditions: chronic inflammation.
Chronic Inflammation: A Hidden Common Denominator
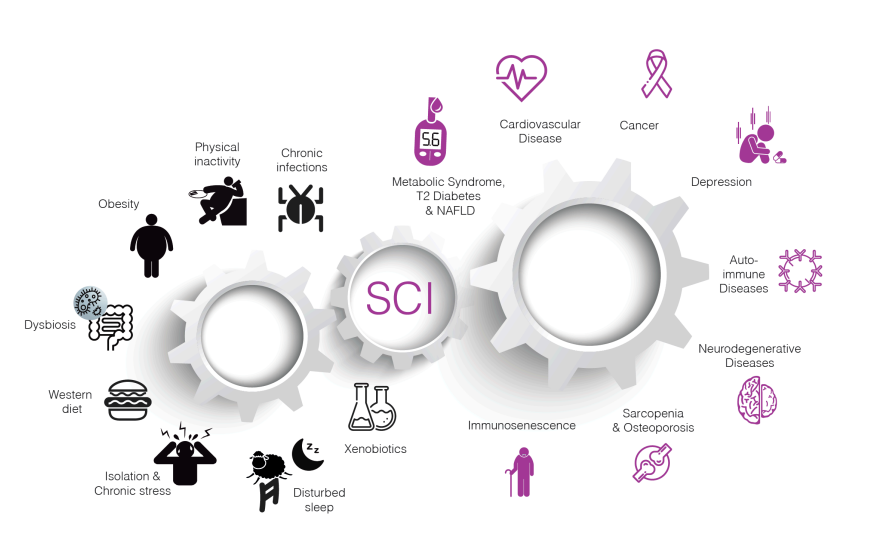
Chronic low-grade inflammation links diverse risk factors to major chronic diseases. Many triggers – such as obesity, physical inactivity, poor diet, smoking, chronic stress, infections, and environmental pollutants – can induce persistent systemic inflammation. This smoldering inflammation in turn contributes to the pathogenesis of cardiometabolic diseases (like metabolic syndrome, type 2 diabetes, fatty liver disease, atherosclerosis), malignancies (cancer), neurodegenerative disorders (Alzheimer’s and other dementias), and autoimmune or degenerative conditions (discovery.ucl.ac.ukdiscovery.ucl.ac.uk).
Unlike the acute inflammation that fights infection or heals injuries, chronic low-grade inflammation is a long-term, dysregulated immune activation. It is now recognized as a key factor in some of the most challenging diseases of our time (nature.com).
In fact, a 2019 cross-disciplinary review in Nature Medicine described systemic chronic inflammation as an accelerant for conditions ranging from cardiovascular disease and cancer to type 2 diabetes and neurodegenerative disease (nature.com). The figure above illustrates how various lifestyle and environmental stressors fuel an inflammatory state, which in turn leads to a constellation of chronic diseases. Notably, type 2 diabetes, malignancies, and Alzheimer’s disease – the very conditions where surrogate markers often mislead – all have significant inflammatory components in their pathogenesis (discovery.ucl.ac.uk).
This connection suggests a reason why “treating the marker” can fail. If an intervention doesn’t address the underlying inflammatory and pathophysiological processes driving the illness, the disease may continue to progress even if the surrogate marker looks better. On the flip side, therapies that directly modulate inflammation or other fundamental disease drivers might improve outcomes even when surrogate markers are unchanged. Below, we delve into specific examples in diabetes, cancer, and Alzheimer’s disease, examining how inflammation and other factors mediate the disconnect between markers and true clinical benefit.
Diabetes: Glucose Control vs Metabolic Disease
Type 2 diabetes mellitus (T2DM) is classically defined by elevated blood glucose, so it’s natural that glucose metrics (fasting glucose, HbA1c) became the primary markers to manage. For decades, the paradigm was “the lower the HbA 1c, the better.”
Drugs were approved solely on their ability to reduce blood sugar. However, a startling lesson came from trials like ACCORD: in high-risk T2DM patients, aggressively pushing HbA1c to near-normal levels (<6%) increased mortality instead of improving it (aafp.orgaafp.org).
In ACCORD, the intensive-therapy group achieved a medianHbA1c of ~6.4% versus 7.5% in the standard group, yet had 19% higher all-cause death rate and more cardiovascular deaths.
The trial was halted early, illustrating that tighter glycemic control is not always better for outcomes – an especially sobering finding given that HbA1c was improved dramatically. Other trials (ADVANCE, VADT) similarly showed no significant reduction in heart attacks or survival from intensive glycemic control in older T2DM patients, and sometimes harm.
The glycemic marker alone was an incomplete picture. In T2DM, underlying drivers include insulin resistance, visceral adiposity, dyslipidemia, and systemic inflammation. Merely forcing blood sugar down (e.g. with high doses of insulin or sulfonylureas) doesn’t reverse these root causes – and might even exacerbate issues like weight gain or hypoglycemia, which carry risks.
Indeed, systemic inflammatory markers like high-sensitivity CRP are often elevated in diabetics and independently predict worse outcomes (mdpi.commdpi.com). Therapies targeting the inflammatory pathways have shown promise. For example, a randomized trial of an IL-1 receptor antagonist (anakinra) in T2DM patients led to improved glycemic control and beta-cell function while also reducing inflammatory cytokines (pubmed.ncbi.nlm.nih.gov). This suggests that quelling inflammation (IL-1 is a key mediator of beta-cell stress) can address the disease process more fundamentally than just chasing blood sugar numbers.
The most impactful new diabetes drugs further support this paradigm shift. GLP-1 receptor agonists (like liraglutide or semaglutide) and SGLT2 inhibitors (like empagliflozin) were initially developed to lower glucose, but they have delivered unexpected benefits in cardiovascular and renal outcomes. Notably, GLP-1 agonists not only lower HbA<sub>1c</sub> and induce weight loss, they also reduce systemic inflammation – meta-analyses confirm that GLP-1 RA therapy significantly lowers CRP levels in diabetics(pubmed.ncbi.nlm.nih.gov).
In major outcome trials (LEADER, SUSTAIN-6, etc.), these drugs cut heart attacks, strokes, and cardiovascular death despite modest differences in HbA<sub>1c</sub>, indicating their benefits stem from improving the overall metabolic/inflammatory milieu. Similarly, SGLT2 inhibitors have revolutionized diabetes care by markedly reducing heart failure and CKD progression – effects attributed to hemodynamic and anti-inflammatory mechanisms (reducing oxidative stress, visceral fat, etc.) rather than glucose lowering per se. In short, treating the whole patient – weight, diet, inflammation, cardiovascular health – works better than treating a glucose number.
The latest guidelines in 2025 reflect this, prioritizing drugs with proven outcome benefits and encouraging lifestyle interventions (diet, exercise) known to reduce inflammatory burden in parallel with improving insulin sensitivity.
Cancer: Tumor Response vs Patient Survival
Oncology has long grappled with the limitations of surrogate endpoints. Drugs are often evaluated on tumor metrics: Does the tumor shrink (objective response rate)? Is tumor growth delayed (progression-free survival, PFS)? These are important signals of drug activity, but the ultimate goal is extending overall survival (OS) and improving quality of life. Unfortunately, many treatments that clear scans or slow tumor growth fail to prolong life. A striking example was bevacizumab for metastatic breast cancer.
Initial trials showed it could add several months to median PFS (by inhibiting tumor angiogenesis), leading to accelerated FDA approval. However, follow-up studies found no significant improvement in OS – patients lived no longer on average, because the cancer would eventually progress and the drug carried toxicity (pmc.ncbi.nlm.nih.gov).
In the pivotal E2100 trial, adding bevacizumab roughly doubled median PFS (11.3 vs 5.8 months) but had no effect on median survival (~26.5 vs 24.8 months, p = n.s.). With risk of serious side effects, the risk-benefit fell apart, and the FDA withdrew the breast cancer indication. This taught oncologists that PFS gains, even if statistically significant, may not translate to real patient benefit – especially if cancer biology or alternate pathways overcome the temporary tumor control.
The bevacizumab case is not isolated. A 2022 review of meta-analyses found that most surrogate endpoints in oncology have only a low-to-moderate correlation with overall survival (pharmacytimes.com). Drugs that “improve” imaging or lab markers might be offering only a short-term or partial fix. Tumors are heterogeneous; knocking back one measurable aspect (like size on a scan or a protein marker level) doesn’t eradicate the disease if resistant clones or metastatic microfoci remain. Here again, the role of inflammation is salient.
Chronic inflammation fosters tumor growth and spread – inflammatory cytokines (IL-1, IL-6, TNFα, etc.) in the tumor microenvironment support angiogenesis, DNA damage, immune evasion, and metastasis. Patients with high inflammatory markers (e.g. CRP or neutrophil-to-lymphocyte ratio) often have worse cancer prognoses (mdpi.commdpi.com). This implies that an intervention narrowly aimed at tumor cells might not succeed if it doesn’t alter the pro-tumor inflammatory milieu or immune response.
Evidence is emerging that targeting inflammation can reduce cancer incidence and improve outcomes. One remarkable finding came from the cardiology realm: the CANTOS trial tested an IL-1β neutralizing antibody (canakinumab) in post-heart-attack patients to prevent recurrent events. As a side benefit, it was observed that lung cancer rates plummeted in the treated group (pubmed.ncbi.nlm.nih.gov).
Over ~3.7 years, lung cancer incidence in patients on high-dose canakinumab was less than half that of placebo (hazard ratio ~0.33) and lung cancer mortality dropped by ~77% (HR 0.23). This was an exploratory finding, but it strongly suggests that blocking a key inflammatory cytokine interrupted nascent lung cancers. Follow-up trials (CANOPY) attempted to use canakinumab to treat established lung cancer; those did not show a survival benefit, underscoring that timing and cancer stage matter – once a tumor is entrenched, inflammation is only one piece of the puzzle (pubmed.ncbi.nlm.nih.gov).
Nonetheless, the idea of cancer chemoprevention via anti-inflammatory agents is gaining traction. Long-term aspirin use is known to lower the risk of certain cancers, especially colorectal cancer. Meta-analyses indicate regular aspirin use is associated with roughly a 25–27% reduction in colorectal cancer incidence over time (journals.lww.com). In Lynch syndrome (a genetic high-risk condition), 2 years of aspirin resulted in significantly fewer colorectal cancers years later (thelancet.com). Aspirin and other NSAIDs likely work by suppressing COX-mediated inflammatory cascades that drive early tumor development in the colon.
In frontline cancer therapy, the advent of immunotherapy (checkpoint inhibitors) has in some ways flipped the script on inflammation – these drugs stimulate an immune attack on tumors, which is acutely inflammatory (often causing autoimmune side effects), yet can lead to durable remissions. They highlight that harnessing the right kind of inflammation (anti-tumor immune response) while suppressing the tumor-promoting chronic inflammation is a delicate balance.
As of 2025, oncology research is increasingly integrative: combining targeted drugs with agents that modulate the microenvironment, metabolism, or immunity. Researchers are exploring, for example, IL-6 inhibitors to combat cancer cachexia and tumor growth, or using low-dose anti-inflammatories to complement immunotherapy without dampening anti-tumor T cells. The key is that success likely requires addressing root drivers – genomic aberrations and the host environment – rather than just shrinking the tumor short-term. Surrogate endpoints remain useful for faster trials, but clinicians have learned to be cautious: an intervention must ultimately prove itself on outcomes that matter to patients (survival, cure rates, quality of life) before being declared a true advance.
Alzheimer’s Disease: Clearing Plaques vs Preserving Cognition
Alzheimer’s disease (AD) presents perhaps the most poignant example of the marker vs disease dilemma. For decades, the dominant hypothesis was that accumulation of beta-amyloid (Aβ) plaques in the brain drives neurodegeneration and dementia. Thus, amyloid levels – as seen on PET scans or measured in cerebrospinal fluid – became key markers, and numerous therapies were designed to reduce amyloid. Many succeeded in lowering amyloid plaques, only to deliver disappointing clinical results.
Monoclonal antibodies like bapineuzumab and solanezumab, and BACE inhibitors that prevent Aβ production, could substantially clear amyloid from the brain, yet patients’ cognitive decline continued unabated. In some cases, cognitive outcomes were no better than placebo, and certain drugs even worsened clinical measures despite reducing plaques (likely due to side effects like inflammation or ARIA brain swelling). These failures forced a reckoning: amyloid is certainly involved in AD, but simply removing the visible plaques does not miraculously restore memory or halt the complex neurodegenerative cascade, especially in moderate to late stages of disease.
The past two years have seen the first glimmers of success for anti-amyloid therapy – drugs like aducanumab and lecanemab earned regulatory approval based on biomarker improvement and a small slowing of cognitive decline in early AD. For instance, lecanemab was shown to reduce brain amyloid load to near-normal levels and to slow the rate of cognitive and functional decline by about 27% over an 18-month trial (nia.nih.goveisai.com).
This is a statistically significant and clinically meaningful slowing, but it’s important to note that patients on lecanemab still experienced decline – the disease course was modestly decelerated, not stopped. In other words, even after “normalizing” the amyloid marker, the underlying neurodegeneration marches on, driven by other factors such as tau tangles, neuronal loss, and chronic neuroinflammation. Indeed, autopsy and biomarker studies show that by the time dementia manifests, a cascade of damage (including tau pathology, synaptic loss, and brain insulin resistance) is well underway.
Inflammation in the brain is a critical mediator here. In AD, the brain’s immune cells (microglia) and inflammatory proteins become activated chronically, creating a toxic environment for neurons. Recent research presented in 2025 has illuminated how Aβ itself can trigger a maladaptive immune response: large amyloid aggregates cause persistent activation of innate immune complexes (myddosomes) in microglia, leading to chronic neuroinflammation unlike the swift, resolving inflammation seen in infectionsnews-medical.net. This chronic inflammation further damages synapses and neurons, and can promote tau pathology – a vicious cycle sustaining the disease.
Given this, it is no surprise that just removing amyloid deposits, while helpful, is insufficient as a cure. We need to address neuroinflammation and other downstream processes. Trials of anti-inflammatory agents in Alzheimer’s have had mixed results so far. For example, epidemiological observations suggested long-term NSAID use might protect against AD, but prospective trials (e.g. using naproxen or ibuprofen in high-risk individuals) did not clearly prevent dementia, possibly because of timing or dosing issues.
Small pilot studies have tested drugs like low-dose colchicine (an inflammasome inhibitor) or hydroxychloroquine in AD, showing they are tolerated and hinting at reduced inflammatory markers, but larger efficacy trials are ongoing (pubmed.ncbi.nlm.nih.gov).
Another approach is targeting immune regulators: genetics has implicated microglial receptors (like TREM2) in AD risk, and therapies to boost microglial cleanup or tamp down their inflammatory overactivation are in development. Additionally, lifestyle interventions that reduce systemic inflammation – e.g. diet (anti-inflammatory Mediterranean or ketogenic diets), exercise, and management of comorbidities – are being studied for their impact on cognitive aging. A recent study even found that an anti-diabetic GLP-1 agonist (liraglutide) slowed brain atrophy in AD, possibly via its anti-inflammatory and neurotrophic effects (nature.com).
The current state of AD research (2025) acknowledges that we must cast a wider net than amyloid. Combination therapy trials are emerging – for instance, pairing an amyloid-clearing antibody with an anti-tau drug or an anti-inflammatory drug to hit multiple targets.
There is also debate about what the best surrogate markers should be in AD. While amyloid PET is a surrogate that correlated with pathology, a truly successful disease-modifying therapy might be better reflected in downstream markers (like phosphorylated tau levels, markers of synaptic integrity, or neuroinflammatory cytokines). Ultimately, cognitive and functional preservation remains the gold standard outcome. The recent anti-amyloid antibodies have taught us both the promise and limitation of focusing on one marker: they validate amyloid as a target (some slowing of decline when removed), but also highlight that Alzheimer’s is more than just amyloid – inflammation, vascular factors, and tau pathology must also be tackled to significantly alter the course of disease.
Conclusion and Future Directions
Across these examples – diabetes, cancer, Alzheimer’s – a clear theme emerges: improving a disease-specific marker does not guarantee improving the disease itself. A pill that lowers blood sugar, shrinks a tumor on scan, or clears amyloid from the brain can still leave patients just as sick or dying just as early if it fails to modify the disease’s underlying pathophysiology.
The literature up to 2025 is replete with humbling trials where the biomarker moved in the hoped-for direction, yet the hard endpoints stayed stubbornly unchanged (or even worsened). This surrogate marker paradox has reshaped how scientists design and evaluate interventions. There is a push for more validated surrogate endpoints – those that truly predict clinical benefit – and for requiring outcome evidence sooner. Researchers like Wallach et al. stress the need for rigorous demonstration that a surrogate is on the causal pathway, not merely correlated (eprints.gla.ac.uk). In practical terms, this means regulatory bodies and clinicians are more cautious in celebrating surrogate wins until patient-centered outcomes are confirmed.
More optimistically, the recognition of inflammation as a common driver is steering innovation toward holistic treatments. Anti-inflammatory therapeutics (e.g. IL-1 or IL-6 inhibitors, colchicine, immune modulators) are being tested in cardiovascular disease, diabetes complications, cancer prevention, and neurodegeneration. Some, like IL-1β blockade and colchicine, have already shown real outcome benefits in cardiovascular trials (mdpi.commdpi.com), and are being repurposed in other fields.
We are also learning the importance of patient selection and timing. An intervention that fails in end-stage disease (when irreversible damage is done) might succeed in early disease or in high-risk but not-yet-ill individuals. For example, clearing amyloid in pre-symptomatic or mild AD might yield bigger cognitive dividends than in moderate AD; likewise, tightly controlling blood sugar or weight very early in the course of diabetes (or pre-diabetes) might prevent complications, whereas doing so late offers little benefit. Thus, precision medicine – getting the right intervention to the right stage and population – is crucial.
In summary, the current state of research underscores that “not everything that counts can be counted” – a normal lab value or imaging result is only as good as the clinical reality behind it. Chronic diseases often involve a network of dysregulation (metabolic, immunologic, genetic) that can’t be captured by a single marker. Physicians and scientists are increasingly aware that treating patients requires going beyond surface metrics to tackle root causes.
In conditions like cancer, diabetes, and Alzheimer’s, this means addressing systemic factors like chronic inflammation, immune balance, metabolic health, and others in tandem with traditional targets. The hope is that by defusing these fundamental drivers, future interventions will align the markers with true disease modification – so that when a biomarker improves, the patient unmistakably improves with it. As we move forward, every proposed therapy will continue to face the key question: does it merely light up the scoreboard, or does it genuinely change the game for patients? The answer must come from rigorous evidence on outcomes that matter, keeping us focused on what ultimately counts – length and quality of human life.
Member discussion